
Push-Pull Effect of Terpyridine Substituted by Triphenylamine Motive—Impact of Viscosity, Polarity and Protonation on Molecular Optical Properties
 , , , and
, , , and
Abstract
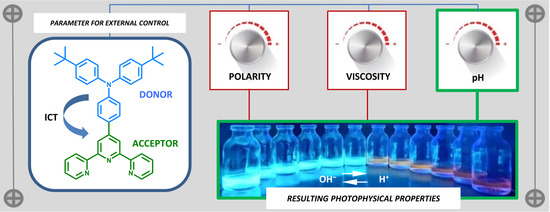
:
1. Introduction
2. Results and Discussion
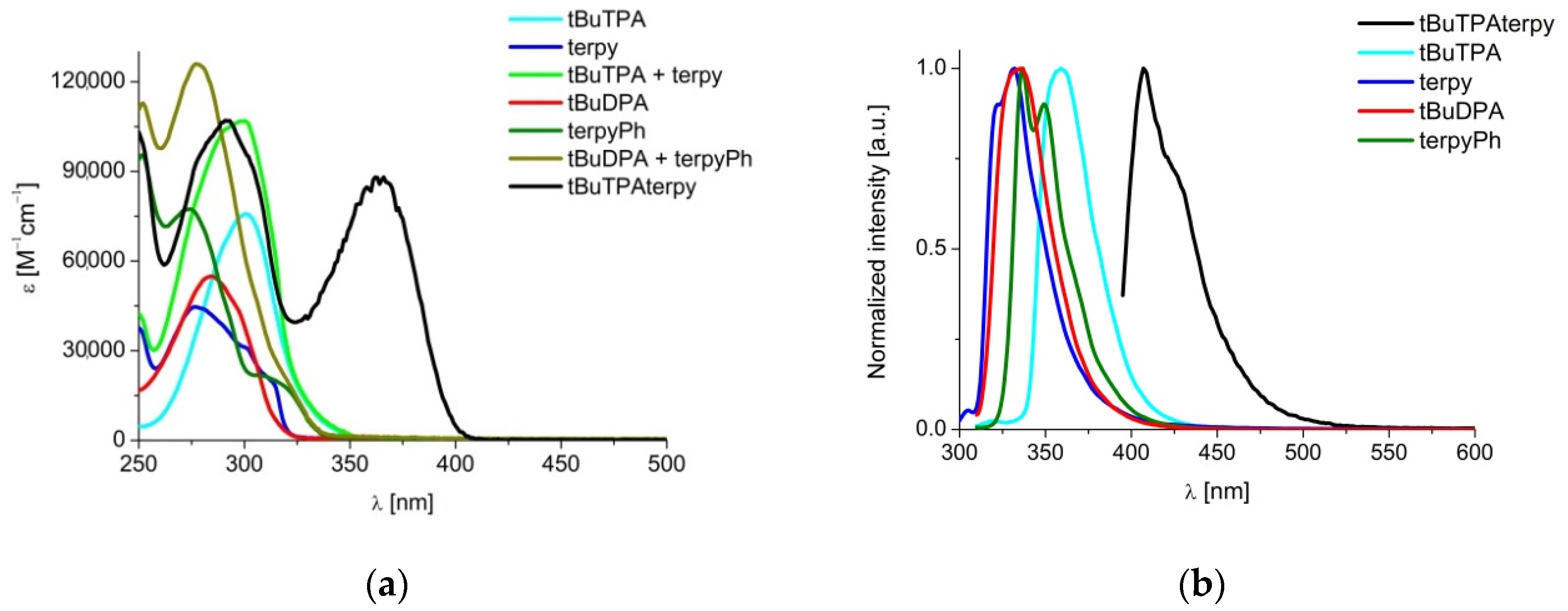
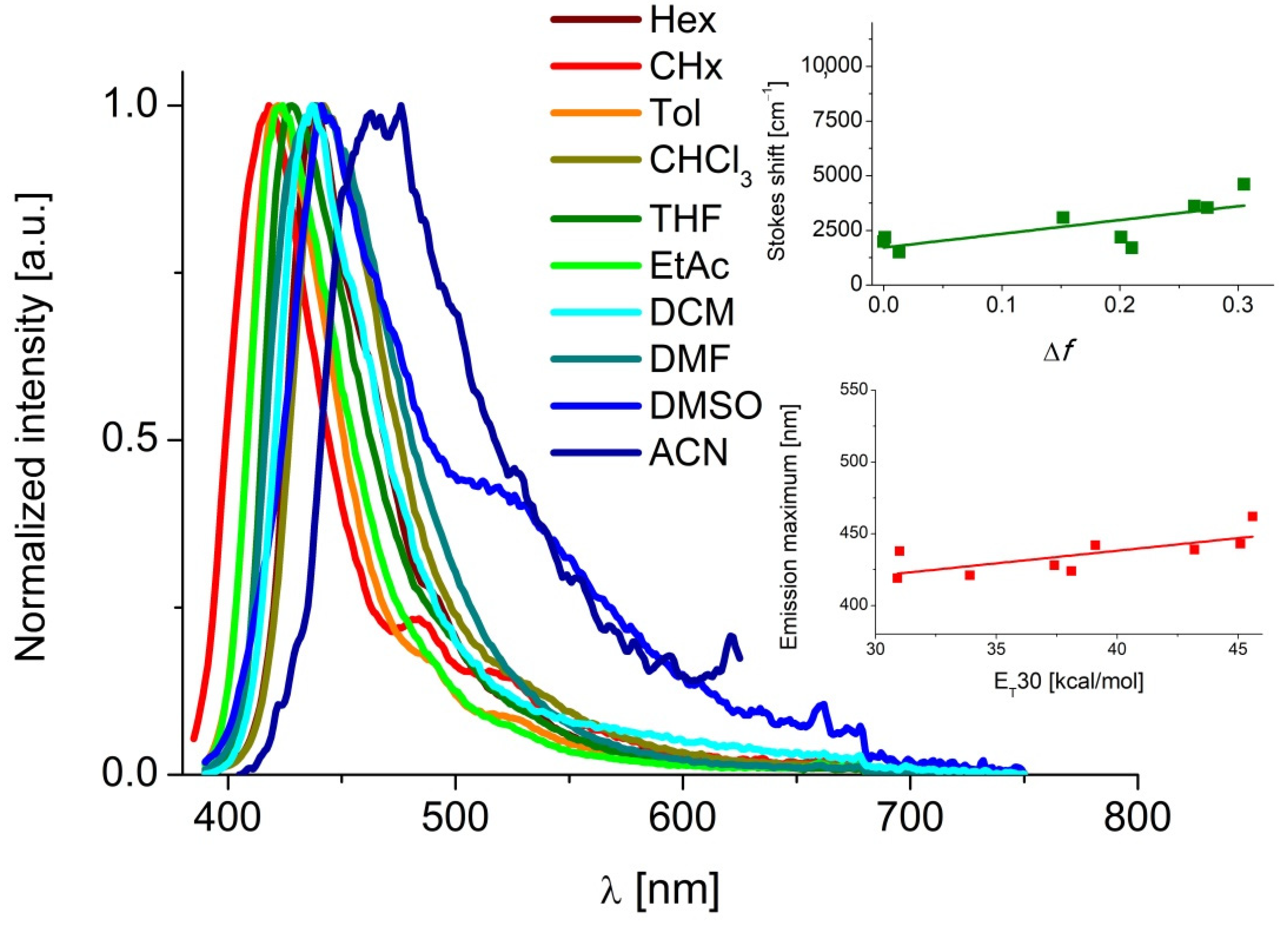
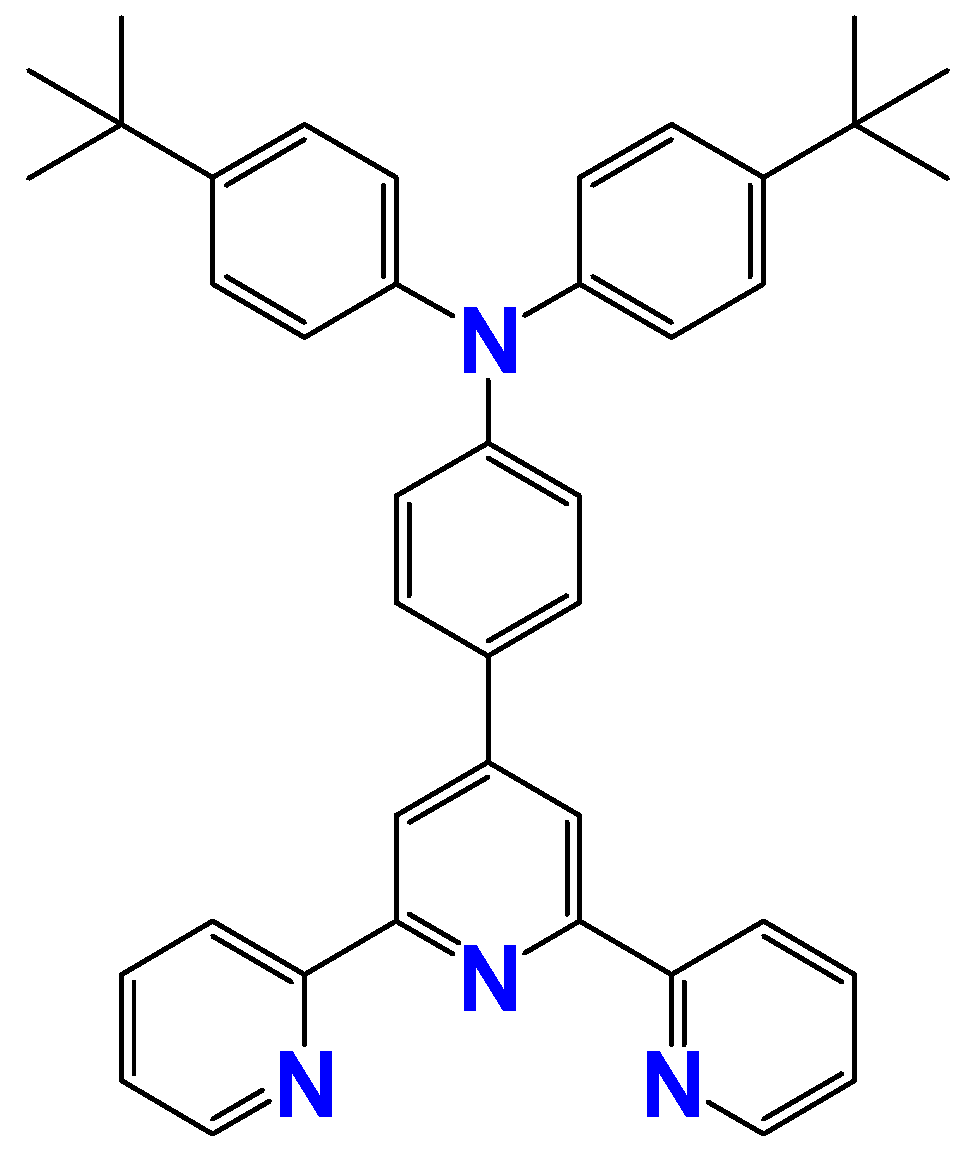
2.1. Optical Spectra of tBuTPAterpy vs Constituent Building Blocks
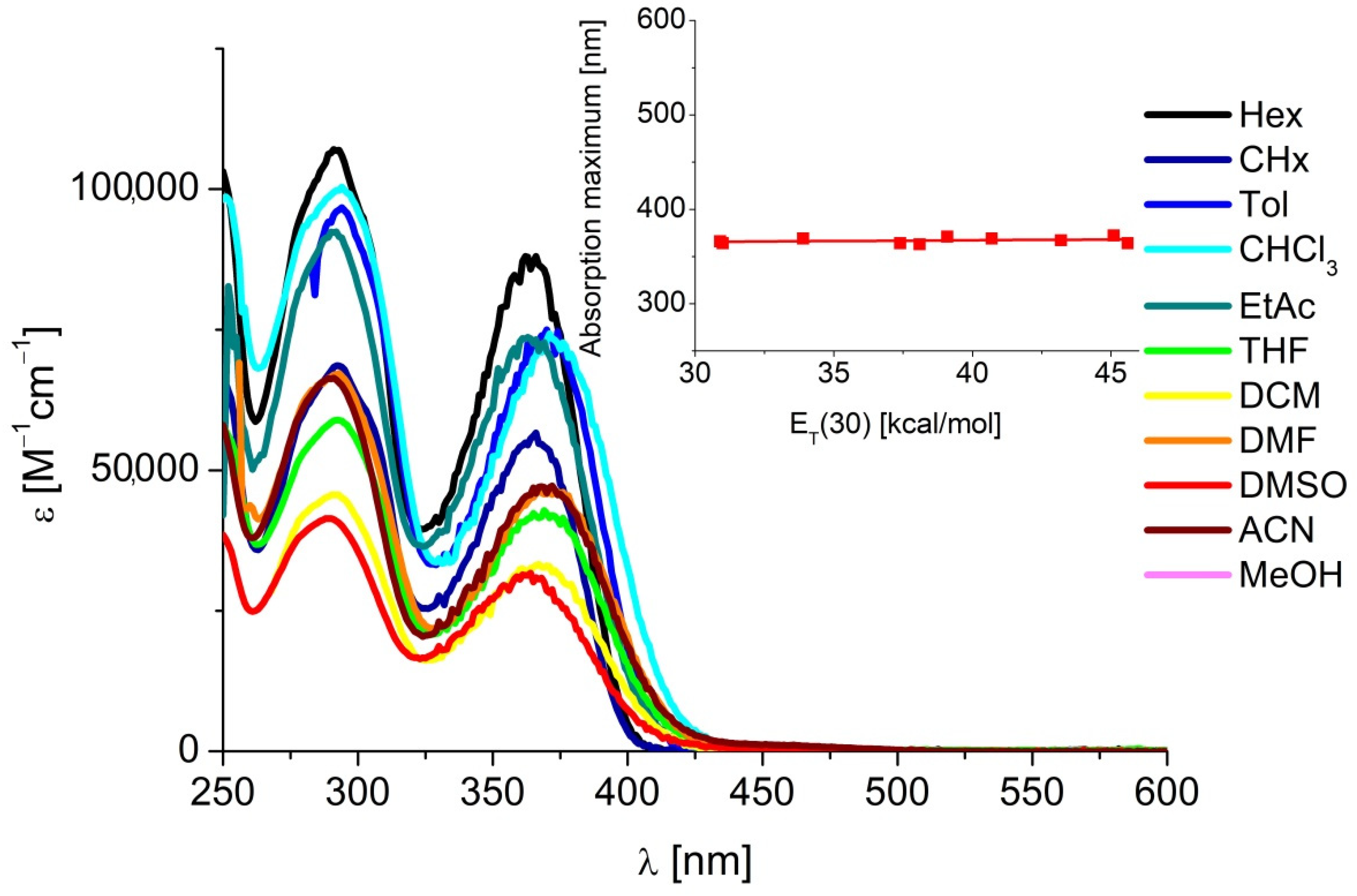
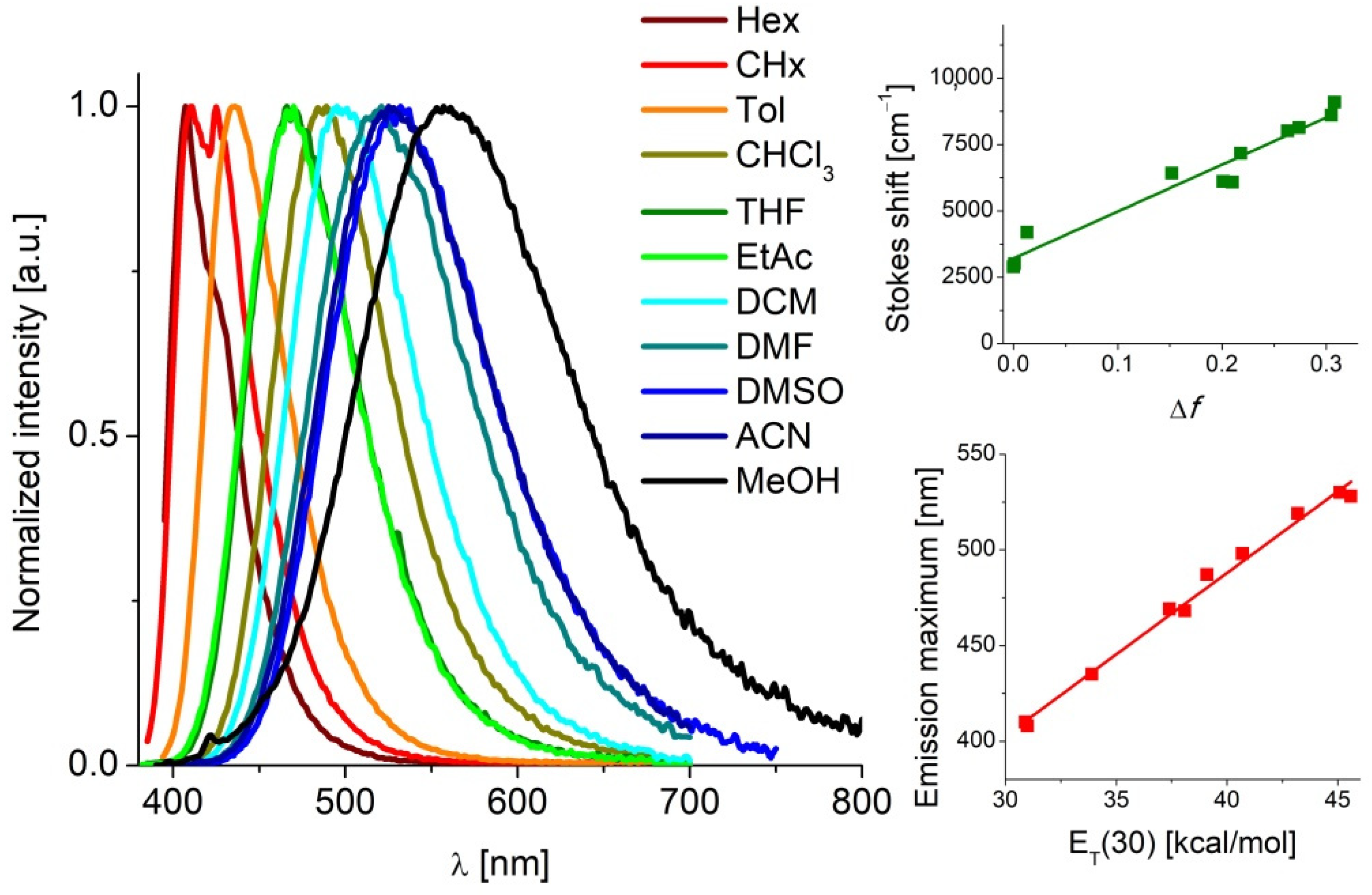
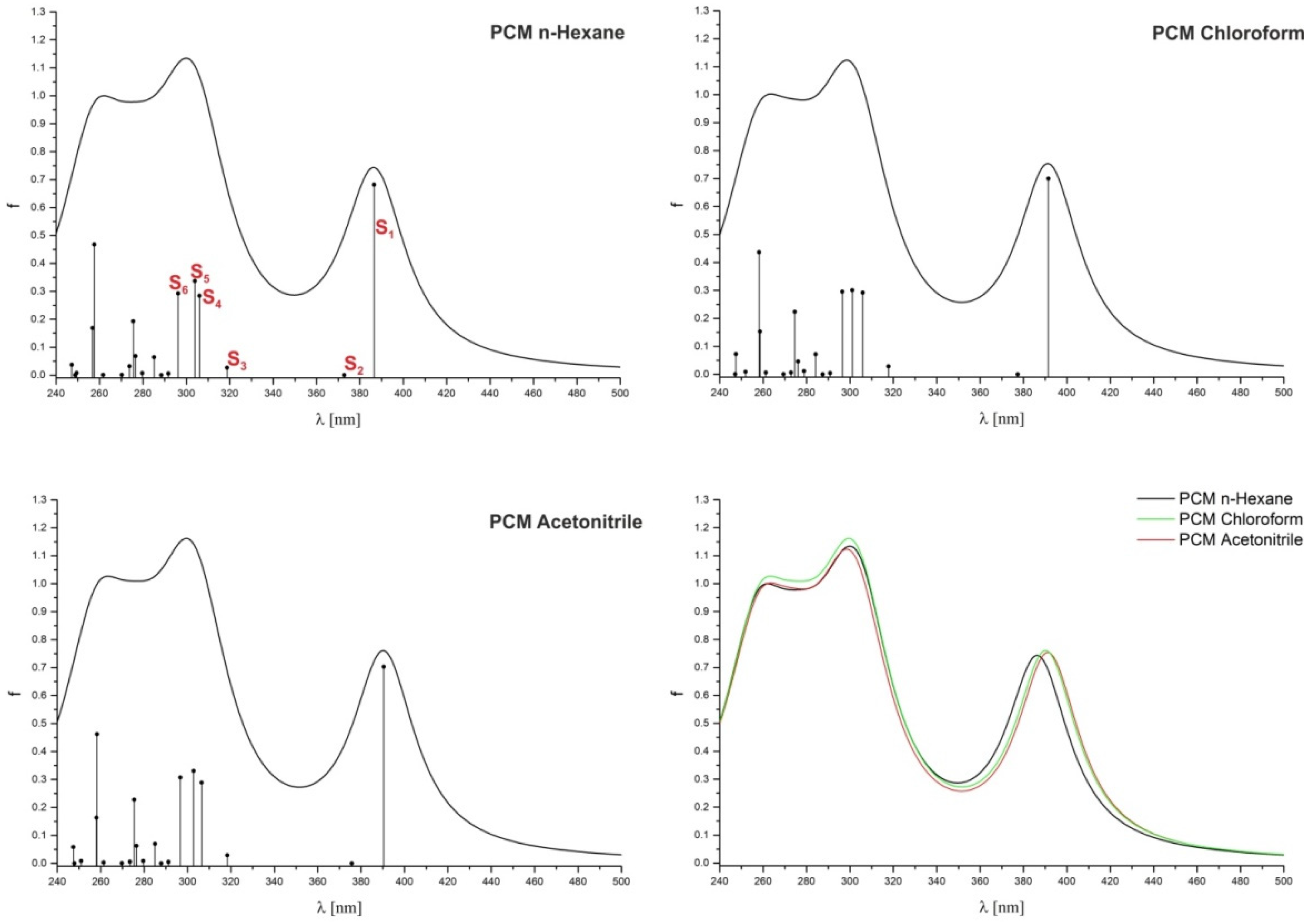
2.2. Solvent Polarity Effect
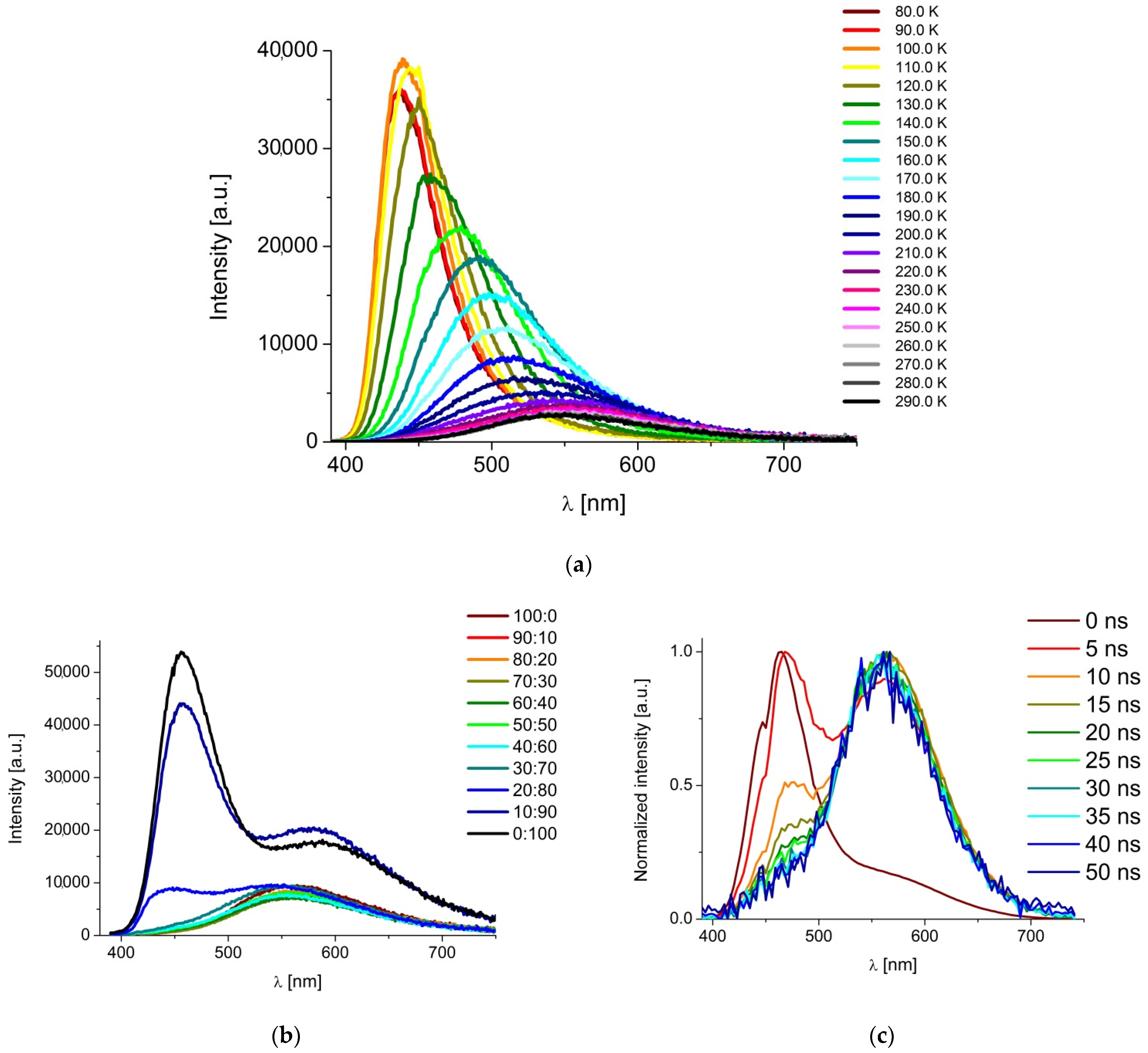
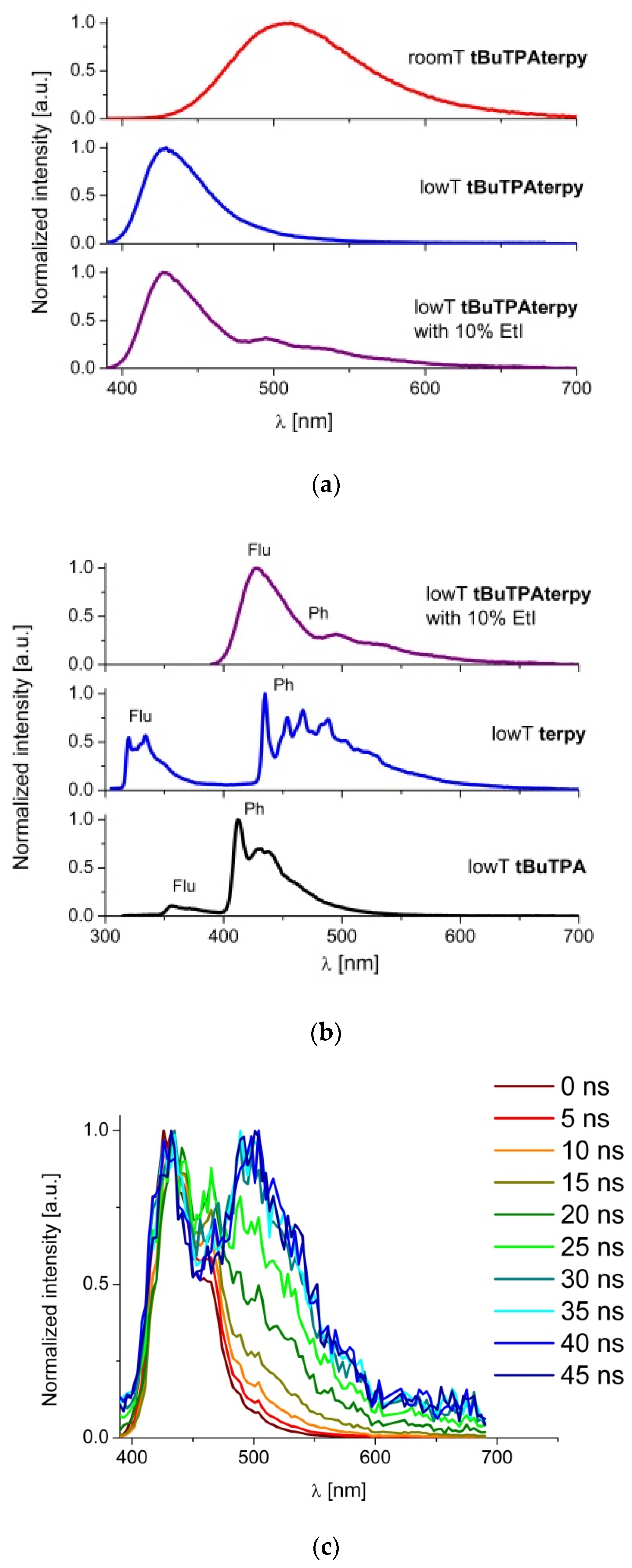
2.3. Temperature and Solvent Viscosity Effects
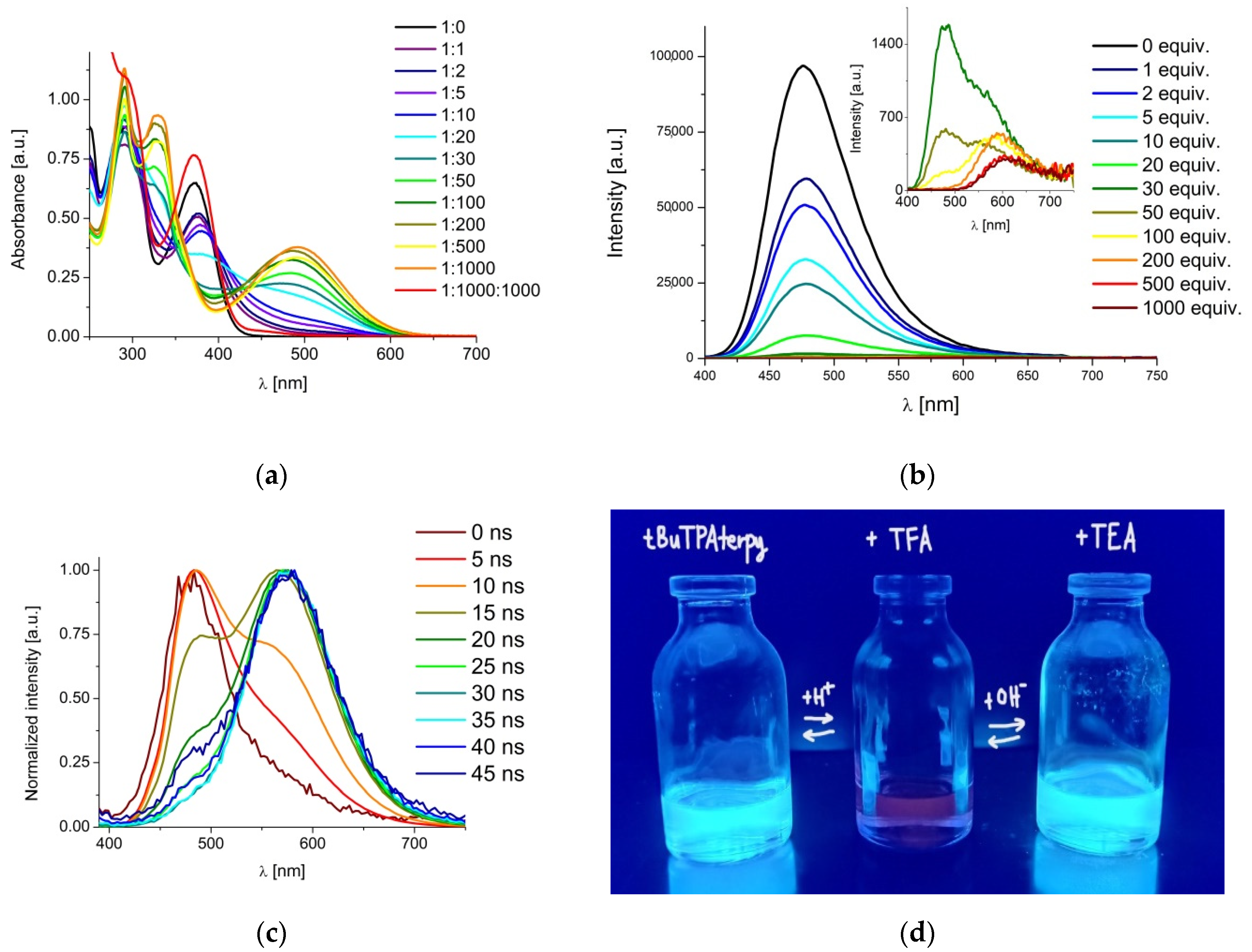
2.4. Protonation Effect
2.5. Phosphorescence of tBuTPAterpy
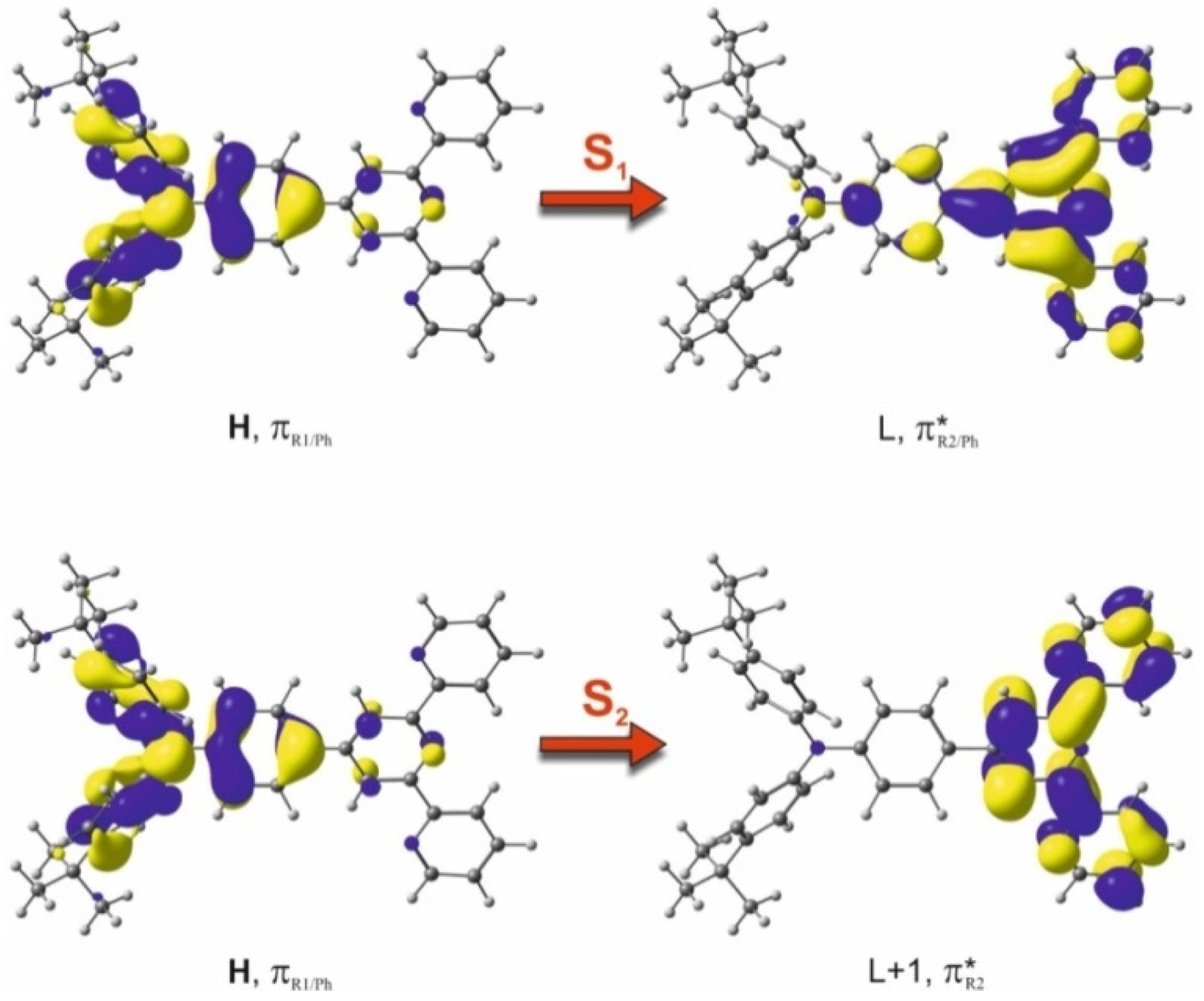
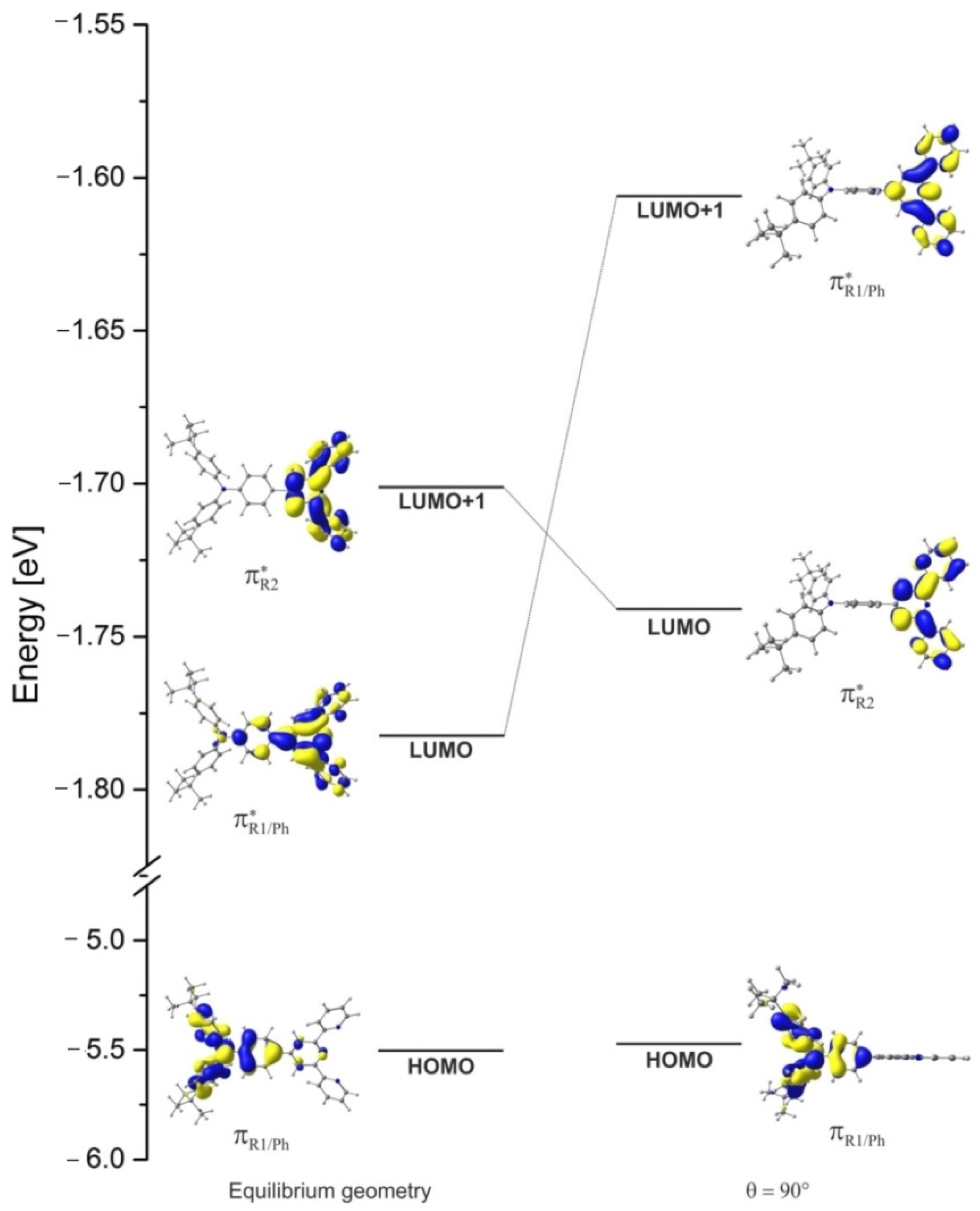
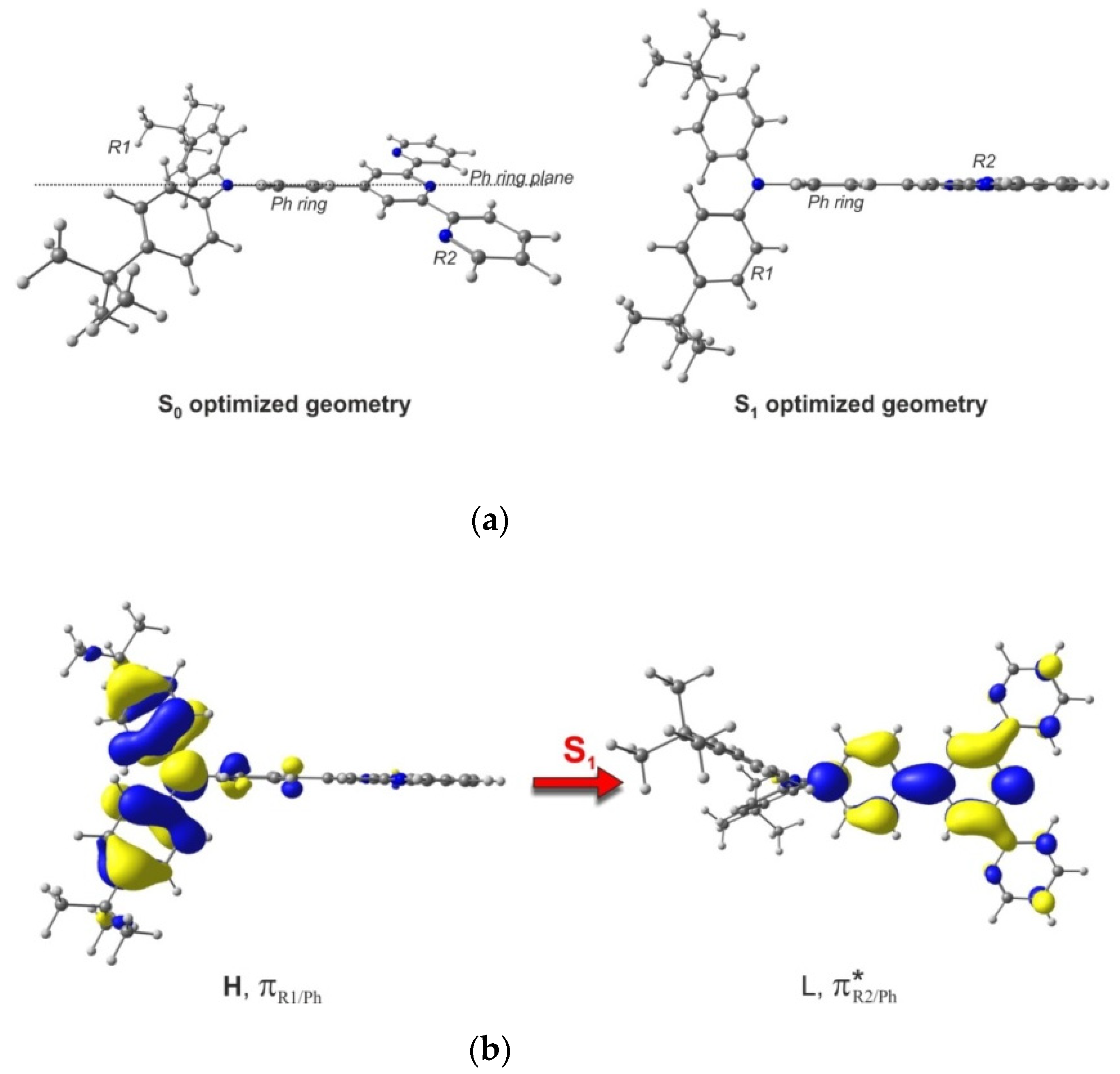
2.6. Quantum Mechanical Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wei, C.; He, Y.; Shi, X.; Song, Z. Terpyridine-Metal Complexes: Applications in Catalysis and Supramolecular Chemistry. Coord. Chem. Rev. 2019, 385, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.T.; Burstall, F.H. 3. Dehydrogenation of Pyridine by Anhydrous Ferric Chloride. J. Chem. Soc. 1932, 1932, 20–30. [Google Scholar] [CrossRef]
- Morgan, G.; Burstall, F.H. 347. Researches on Residual Affinity and Co-Ordination. Part XXXVII. Complex Metallic Salts Containing 2:6-Di-2′-Pyridylpyridine (2:2′:2″-Tripyridyl). J. Chem. Soc. 1937, 1937, 1649–1655. [Google Scholar] [CrossRef]
- Winter, A.; Schubert, U.S. Metal-Terpyridine Complexes in Catalytic Application—A Spotlight on the Last Decade. ChemCatChem 2020, 12, 2890–2941. [Google Scholar] [CrossRef]
- Agosti, A.; Kuna, E.; Bergamini, G. Divergent Terpyridine-Based Coordination for the Construction of Photoactive Supramolecular Structures. Eur. J. Inorg. Chem. 2019, 2019, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Han, Y.; Gao, Z.; Wang, F. Multicomponent Assembled Systems Based on Platinum(II) Terpyridine Complexes. Acc. Chem. Res. 2018, 51, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Mede, T.; Jäger, M.; Schubert, U.S. “Chemistry-on-the-Complex”: Functional RuII Polypyridyl-Type Sensitizers as Divergent Building Blocks. Chem. Soc. Rev. 2018, 47, 7577–7627. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Newkome, G.R. Terpyridine-Based Metallosupramolecular Constructs: Tailored Monomers to Precise 2D-Motifs and 3D-Metallocages. Chem. Soc. Rev. 2018, 47, 3991–4016. [Google Scholar] [CrossRef]
- Attwood, M.; Turner, S.S. Back to Back 2,6-Bis(Pyrazol-1-Yl)Pyridine and 2,2′:6′,2″-Terpyridine Ligands: Untapped Potential for Spin Crossover Research and Beyond. Coord. Chem. Rev. 2017, 353, 247–277. [Google Scholar] [CrossRef]
- Heinemann, F.; Karges, J.; Gasser, G. Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res. 2017, 50, 2727–2736. [Google Scholar] [CrossRef]
- Chi, Y.; Chang, T.-K.; Ganesan, P.; Rajakannu, P. Emissive Bis-Tridentate Ir(III) Metal Complexes: Tactics, Photophysics and Applications. Coord. Chem. Rev. 2017, 346, 91–100. [Google Scholar] [CrossRef]
- Saccone, D.; Magistris, C.; Barbero, N.; Quagliotto, P.; Barolo, C.; Viscardi, G. Terpyridine and Quaterpyridine Complexes as Sensitizers for Photovoltaic Applications. Materials 2016, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Yan, Y.; Helbig, B.J.; Huang, Z.; Lian, T.; Schmehl, R.H. The Influence of Ligand Localized Excited States on the Photophysics of Second Row and Third Row Transition Metal Terpyridyl Complexes: Recent Examples and a Case Study. Coord. Chem. Rev. 2015, 282–283, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Adeloye, A.O.; Ajibade, P.A. Towards the Development of Functionalized PolypyridineLigands for Ru(II) Complexes as Photosensitizers InDye-Sensitized Solar Cells (DSSCs). Molecules 2014, 19, 12421–12460. [Google Scholar] [CrossRef] [Green Version]
- Winter, A.; Gottschaldt, M.; Newkome, G.R.; Schubert, U.S. Terpyridines and Their Complexes with First Row Transition Metal Ions:Cytotoxicity, Nuclease Activity and Self-Assembly of Biomacromolecules. Curr. Top. Med. Chem. 2012, 12, 158–175. [Google Scholar] [CrossRef]
- Wild, A.; Winter, A.; Schlütter, F.; Schubert, U.S. Advances in the Field of π-Conjugated 2,2′:6′,2″-Terpyridines. Chem. Soc. Rev. 2011, 40, 1459–1511. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Hager, M.D.; Newkome, G.R.; Schubert, U.S. The Marriage of Terpyridines and Inorganic Nanoparticles: Synthetic Aspects, Characterization Techniques, and Potential Applications. Adv. Mater. 2011, 23, 5728–5748. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.D. Platinum Complexes of Terpyridine: Synthesis, Structure and Reactivity. Coord. Chem. Rev. 2009, 253, 449–478. [Google Scholar] [CrossRef]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-Planar Pd(II), Pt(II), and Au(III) Terpyridine Complexes: Their Syntheses, Physical Properties, Supramolecular Constructs, and Biomedical Activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Kanaizuka, K.; Nishimori, Y.; Yamanoi, Y. Construction of Redox- and Photo-Functional Molecular Systems on Electrode Surface for Application to Molecular Devices. Coord. Chem. Rev. 2007, 251, 2674–2687. [Google Scholar] [CrossRef]
- Pal, A.K.; Hanan, G.S. Design, Synthesis and Excited-State Properties of Mononuclear Ru(II) Complexes of Tridentate Heterocyclic Ligands. Chem. Soc. Rev. 2014, 43, 6184–6197. [Google Scholar] [CrossRef]
- Siebert, R.; Akimov, D.; Schmitt, M.; Winter, A.; Schubert, U.S.; Dietzek, B.; Popp, J. Spectroscopic Investigation of the Ultrafast Photoinduced Dynamics in π-Conjugated Terpyridines. ChemPhysChem 2009, 10, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.-Q.; Bourget-Merle, L.; Polson, M.I.J.; Hanan, G.S.; Juris, A.; Loiseau, F.; Campagna, S. The Multichromophore Approach: Prolonged Room-Temperature Luminescence Lifetimes in RuII Complexes Based on Tridentate Polypyridine Ligands. Chem. A Eur. J. 2006, 12, 8539–8548. [Google Scholar] [CrossRef] [PubMed]
- Castellano, F.N.; Pomestchenko, I.E.; Shikhova, E.; Hua, F.; Muro, M.L.; Rajapakse, N. Photophysics in Bipyridyl and Terpyridyl Platinum(II) Acetylides. Coord. Chem. Rev. 2006, 250, 1819–1828. [Google Scholar] [CrossRef]
- Medlycott, E.A.; Hanan, G.S. Designing Tridentate Ligands for Ruthenium(II) Complexes with Prolonged Room Temperature Luminescence Lifetimes. Chem. Soc. Rev. 2005, 34, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hanan, G.S.; Loiseau, F.; Campagna, S. Prolonged Luminescence Lifetimes of Ru(II) Complexes via the Multichromophore Approach: The Excited-State Storage Element Can Be on a Ligand Not Involved in the MLCT Emitting State. Chem. Commun. 2004, 18, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Palion-Gazda, J.; Machura, B.; Klemens, T.; Szlapa-Kula, A.; Krompiec, S.; Siwy, M.; Janeczek, H.; Schab-Balcerzak, E.; Grzelak, J.; Maćkowski, S. Structure-Dependent and Environment-Responsive Optical Properties of the Trisheterocyclic Systems with Electron Donating Amino Groups. Dye. Pigment. 2019, 166, 283–300. [Google Scholar] [CrossRef]
- Fernández-Terán, R.; Sévery, L. Living Long and Prosperous: Productive Intraligand Charge-Transfer States from a Rhenium(I) Terpyridine Photosensitizer with Enhanced Light Absorption. Inorg. Chem. 2021, 60, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G. Recent Advances in Twisted Intramolecular Charge Transfer (TICT) Fluorescence and Related Phenomena in Materials Chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.J.; Kolanowski, J.L.; Filipek, W.K.; Lim, Z.; Moore, E.G.; Stagni, S.; New, E.J.; Massi, M. Versatility of Terpyridine-Functionalised Aryl Tetrazoles: Photophysical Properties, Ratiometric Sensing of Zinc Cations and Sensitisation of Lanthanide Luminescence. Eur. J. Inorg. Chem. 2017, 2017, 5260–5270. [Google Scholar] [CrossRef]
- Achelle, S.; Rodríguez-López, J.; Katan, C.; Robin-le Guen, F. Luminescence Behavior of Protonated Methoxy-Substituted Diazine Derivatives: Toward White Light Emission. J. Phys. Chem. C 2016, 120, 26986–26995. [Google Scholar] [CrossRef] [Green Version]
- Wałęsa-Chorab, M.; Tremblay, M.-H.; Ettaoussi, M.; Skene, W.G. Photophysical, Electrochemical, and Spectroelectrochemical Investigation of Electronic Push–Pull Benzothiadiazole Fluorophores. Pure Appl. Chem. 2015, 87, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.; Kakkar, R.; Bishnoi, S.; Milton, M.D. Synthesis of Acceptor-Donor-Acceptor Based Phenothiazine-5-Oxide Aldehydes Displaying Large Stokes Shift- “on-off-on” Acidofluorochromic Switch and Molecular Logic Gate Operation. J. Photochem. Photobiol. A Chem. 2022, 430, 113944. [Google Scholar] [CrossRef]
- Chaudhary, S.; Mukherjee, M.; Paul, T.K.; Taraphder, S.; Milton, M.D. Novel Thiazoline-Phenothiazine Based “Push-Pull” Molecules as Fluorescent Probes for Volatile Acids Detection. J. Photochem. Photobiol. A Chem. 2020, 397, 112509. [Google Scholar] [CrossRef]
- Mishra, A.; Chaterjee, S.; Krishnamoorthy, G. Intramolecular Charge Transfer Emission of Trans-2-[4′-(Dimethylamino)Styryl]Benzimidazole: Effect of Solvent and PH. J. Photochem. Photobiol. A Chem. 2013, 260, 50–58. [Google Scholar] [CrossRef]
- Sachdeva, T.; Milton, M.D. Novel Push-Pull Based Phenothiazine-Benzothiazole Derivatives Integrated with Molecular Logic Gate Operation for Reversible Volatile Acid Detection. J. Mol. Struct. 2021, 1243, 130768. [Google Scholar] [CrossRef]
- Gao, A.; Han, Q.; Wang, Q.; Cao, X.; Chang, X.; Zhou, Y. Triphenylamine Derivative-Based Supramolecular Self-Assembly System for Selective Sensing Methanol via Hydrogen Bonding. Dye. Pigment. 2021, 195, 109689. [Google Scholar] [CrossRef]
- Song, P.; Sun, S.-G.; Liu, J.-Y.; Xu, Y.-Q.; Han, K.-L.; Peng, X.-J. Theoretical and Experimental Study on the Intramolecular Charge Transfer Excited State of the New Highly Fluorescent Terpyridine Compound. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 74, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Peng, M.; Shen, H.; Wang, Y.; Zhang, Q.; Wang, H.; Zhang, J.; Zhou, H.; Yang, J.; Wu, J.; et al. A Novel D-A Type Terpyridine-Based Carbazole Zn(II) Complex with Enhanced Two-Photon Absorption and Its Bioimaging Application. Dye. Pigment. 2015, 120, 328–334. [Google Scholar] [CrossRef]
- Bastug, E.; Kursunlu, A.N.; Guler, E. A Fluorescent Clever Macrocycle: Deca-Bodipy Bearing a Pillar [5]Arene and Its Selective Binding of Asparagine in Half-Aqueous Medium. J. Lumin. 2020, 225, 117343. [Google Scholar] [CrossRef]
- Kursunlu, A.N. Synthesis and Photophysical Properties of Modifiable Single, Dual, and Triple-Boron Dipyrromethene (Bodipy) Complexes. Tetrahedron Lett. 2015, 56, 1873–1877. [Google Scholar] [CrossRef]
- Liang, J.; Zhu, C.; Cao, Z. Electronic and Optical Properties of the Triphenylamine-Based Organic Dye Sensitized TiO2 Semiconductor: Insight from First Principles Calculations. Phys. Chem. Chem. Phys. 2013, 15, 13844–13851. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.D.; Tathe, A.B.; Padalkar, V.S.; Umape, P.G.; Sekar, N. Red Emitting Solid State Fluorescent Triphenylamine Dyes: Synthesis, Photo-Physical Property and DFT Study. Dye. Pigment. 2013, 97, 429–439. [Google Scholar] [CrossRef]
- Wang, G.; Hu, Y.; Chen, Y.; Liao, X.; Li, Z.; Chen, X.; Wang, X.; Liu, B. Effect of Multidonor and Insertion Position of a Chromophore on the Photovoltaic Properties of Phenoxazine Dyes. ACS Omega 2020, 5, 22621–22630. [Google Scholar] [CrossRef]
- Heng, P.; An, B.; Ren, H.; Hu, Y.; Guo, X.; Mao, L.; Wang, L.; Zhang, J. Influence of Different Molecular Design Strategies on Photovoltaic Properties of a Series of Triphenylamine-Based Organic Dyes for Dye-Sensitized Solar Cells: Insights from Theoretical Investigations. J. Phys. Chem. C 2020, 124, 15036–15044. [Google Scholar] [CrossRef]
- Liu, J.; Luo, Y.; Li, L.; Wang, G.; Wang, X.; Chen, Y.; Liu, B. Photovoltaic Performance of 4,8-Bis(2′-Ethylhexylthiophene)Thieno[2,3-f]Benzofuran-Based Dyes Fabricated with Different Donors in Dye-Sensitized Solar Cells. ACS Omega 2020, 5, 12440–12450. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, M.; Toufik, H.; Bouzzine, S.M.; Lamchouri, F. Bridge Effect on the Charge Transfer and Optoelectronic Properties of Triphenylamine-Based Organic Dye Sensitized Solar Cells: Theoretical Approach. Res. Chem. Intermed. 2020, 46, 3961–3978. [Google Scholar] [CrossRef]
- Kumavat, P.P.; Sonar, P.; Dalal, D.S. An Overview on Basics of Organic and Dye Sensitized Solar Cells, Their Mechanism and Recent Improvements. Renew. Sustain. Energy Rev. 2017, 78, 1262–1287. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, A. Triphenylamine Based Dyes for Dye Sensitized Solar Cells: A Review. Sol. Energy 2016, 123, 127–144. [Google Scholar] [CrossRef]
- Kursunlu, A.N.; Baslak, C. A Bodipy-Bearing Pillar[5]Arene for Mimicking Photosynthesis: Multi-Fluorophoric Light Harvesting System. Tetrahedron Lett. 2018, 59, 1958–1962. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, H.; Wang, Z.; Qin, A.; Tang, B.Z. Planarized Intramolecular Charge Transfer on Triphenylamine-Modified Pyrazine and Its Application in Organic Light-Emitting Diodes. J. Mater. Chem. C 2020, 8, 4754–4762. [Google Scholar] [CrossRef]
- Braveenth, R.; Jung, H.; Kim, K.; Kim, B.M.; Bae, I.-J.; Kim, M.; Chai, K.Y. Fluorene–Triphenylamine-Based Bipolar Materials: Fluorescent Emitter and Host for Yellow Phosphorescent OLEDs. Appl. Sci. 2020, 10, 519. [Google Scholar] [CrossRef] [Green Version]
- Bian, M.; Zhang, D.; Wang, Y.; Chung, Y.-H.; Liu, Y.; Ting, H.; Duan, L.; Chen, Z.; Bian, Z.; Liu, Z.; et al. Long-Lived and Highly Efficient TADF-PhOLED with “(A)n–D–(A)n” Structured Terpyridine Electron-Transporting Material. Adv. Funct. Mater. 2018, 28, 1800429. [Google Scholar] [CrossRef]
- Sasabe, H.; Hayasaka, Y.; Komatsu, R.; Nakao, K.; Kido, J. Highly Luminescent π-Conjugated Terpyridine Derivatives Exhibiting Thermally Activated Delayed Fluorescence. Chem. A Eur. J. 2017, 23, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Zhang, H.; Xu, J.; Wen, Y.; Zhang, J.; Liu, H.; Yao, Y.; Zhang, Z. Development of Solution-Dispersible Hyperbranched Conjugated Polymer Nanoparticles for Fe3+ Fluorescent Detection and Their Application in Logic Gate. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3694–3700. [Google Scholar] [CrossRef]
- Dheepika, R.; Shaji, A.; Imran, P.M.; Nagarajan, S. Improving Device Performance of P-Type Organic Field-Effect Transistor Using Butterfly like Triarylamines. Org. Electron. 2020, 81, 105568. [Google Scholar] [CrossRef]
- Dheepika, R.; Abhijnakrishna, R.; Imran, P.M.; Nagarajan, S. High Performance P-Channel and Ambipolar OFETs Based on Imidazo[4,5-f]-1,10-Phenanthroline-Triarylamines. RSC Adv. 2020, 10, 13043–13049. [Google Scholar] [CrossRef] [Green Version]
- Dheepika, R.; Mohamed Imran, P.; Bhuvanesh, N.S.P.; Nagarajan, S. Solution-Processable Unsymmetrical Triarylamines: Towards High Mobility and ON/OFF Ratio in Bottom-Gated OFETs. Chem. A Eur. J. 2019, 25, 15155–15163. [Google Scholar] [CrossRef]
- Bathula, C.; Appiagyei, A.B.; Yadav, H.; K., A.K.; Ramesh, S.; Shrestha, N.K.; Shinde, S.; Kim, H.-S.; Kim, H.S.; Reddy, L.V.; et al. Facile Synthesis of Triphenylamine Based Hyperbranched Polymer for Organic Field Effect Transistors. Nanomaterials 2019, 9, 1787. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhu, X.; Zhang, J.; Wang, H.; Liu, G.; Bu, Y.; Yu, J.; Tian, Y.; Zhou, H. AIE-Based Theranostic Agent: In Situ Tracking Mitophagy Prior to Late Apoptosis To Guide the Photodynamic Therapy. ACS Appl. Mater. Interfaces 2020, 12, 1988–1996. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, X.; Zhang, H.; Ou, H.; Lam, J.W.Y.; Liu, Y.; Shi, L.; Ding, D.; Tang, B.Z. Molecular Motion in Aggregates: Manipulating TICT for Boosting Photothermal Theranostics. J. Am. Chem. Soc. 2019, 141, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liang, P.; Tang, Q.; Yang, X.; Si, W.; Huang, W.; Zhang, Q.; Dong, X. Diketopyrrolopyrrole–Triphenylamine Organic Nanoparticles as Multifunctional Reagents for Photoacoustic Imaging-Guided Photodynamic/Photothermal Synergistic Tumor Therapy. ACS Nano 2017, 11, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Maroń, A.M.; Szlapa-Kula, A.; Matussek, M.; Kruszynski, R.; Siwy, M.; Janeczek, H.; Grzelak, J.; Maćkowski, S.; Schab-Balcerzak, E.; Machura, B. Photoluminescence Enhancement of Re(I) Carbonyl Complexes Bearing D–A and D–π–A Ligands. Dalton Trans. 2020, 49, 4441–4453. [Google Scholar] [CrossRef] [PubMed]
- Jarusuwannapoom, T.; Hongrojjanawiwat, W.; Jitjaicham, S.; Wannatong, L.; Nithitanakul, M.; Pattamaprom, C.; Koombhongse, P.; Rangkupan, R.; Supaphol, P. Effect of Solvents on Electro-Spinnability of Polystyrene Solutions and Morphological Appearance of Resulting Electrospun Polystyrene Fibers. Eur. Polym. J. 2005, 41, 409–421. [Google Scholar] [CrossRef]
- Dong, T.; Knoshaug, E.P.; Pienkos, P.T.; Laurens, L.M.L. Lipid Recovery from Wet Oleaginous Microbial Biomass for Biofuel Production: A Critical Review. Appl. Energy 2016, 177, 879–895. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.S.; Rana, D. Preparation of Some Novel Copper(I) Complexes and Their Molar Conductances in Organic Solvents. Z. Für Nat. A 2009, 64, 269–272. [Google Scholar] [CrossRef]
- Tunuli, M.S.; Rauf, M.A. Farhataziz Dimroth’s ET(30) as Parameters of Solvent Polarity: A Caveat. J. Photochem. 1984, 24, 411–413. [Google Scholar] [CrossRef]
- Yang, Z.; Qin, W.; Lam, J.W.Y.; Chen, S.; Sung, H.H.Y.; Williams, I.D.; Tang, B.Z. Fluorescent PH Sensor Constructed from a Heteroatom-Containing Luminogen with Tunable AIE and ICT Characteristics. Chem. Sci. 2013, 4, 3725–3730. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.; Mandal, A.; Mukhopadhyay, M.; Maity, D.K.; Bhattacharyya, S.P. Spectral Signatures of Intramolecular Charge Transfer Process in β-Enaminones: A Combined Experimental and Theoretical Analysis. J. Phys. Chem. B 2009, 113, 10779–10791. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, X.; Ma, R.; Kuang, Z.; Guo, Q.; Xia, A. Intramolecular Charge Transfer and Solvation of Photoactive Molecules with Conjugated Push–Pull Structures. ChemPhysChem 2016, 17, 3245–3251. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, W.; Liu, Z.; Meng, L.; Bai, B.; Wang, H.; Li, M. Enhancement of Intramolecular Charge Transfer Strength in Diphenylamine Substituted Symmetric 1,3,4-Oxadiazole Derivatives. RSC Adv. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khopkar, S.; Jachak, M.; Shankarling, G. Viscosity Sensitive Semisquaraines Based on 1, 1, 2-Trimethyl-1H-Benzo[e]Indole: Photophysical Properties, Intramolecular Charge Transfer, Solvatochromism, Electrochemical and DFT Study. J. Mol. Liq. 2019, 285, 123–135. [Google Scholar] [CrossRef]
- Lippert, E. Dipolmoment Und Elektronenstruktur von Angeregten Molekülen. Z. Für Nat. A 1955, 10, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Kanaparthi, R.K.; Saha, S.; Singh, M.; Akhila, M. Photophysical Properties of 4-(Dicyanomethylene)-2-Methyl-6-(4-Dimethylaminostyryl)-4H-Pyran (DCM) and Optical Sensing Applications; IntechOpen: London, UK, 2020; ISBN 978-1-83968-224-7. [Google Scholar]
- Baschieri, A.; Sambri, L.; Gualandi, I.; Tonelli, D.; Monti, F.; Esposti, A.D.; Armaroli, N. Carbazole-Terpyridine Donor–Acceptor Luminophores. RSC Adv. 2013, 3, 6507–6517. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Yamabe, S.; Kanehisa, N.; Inoue, T.; Takashima, H. Syntheses, X-ray Crystal Structures, and Emission Properties of Diprotonated Tetrapyridylpyrazine and Triprotonated Terpyridine. J. Phys. Org. Chem. 2016, 29, 269–275. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Yamabe, S.; Kanehisa, N.; Takashima, H.; Tsukahara, K. A Metal Free Blue Emission by the Protonated 2,2′:6′,2″-Terpyridine Hexafluorophosphate. J. Phys. Org. Chem. 2009, 22, 410–417. [Google Scholar] [CrossRef]
- Taniya, O.S.; Fedotov, V.V.; Novikov, A.S.; Sadieva, L.K.; Krinochkin, A.P.; Kovalev, I.S.; Kopchuk, D.S.; Zyryanov, G.V.; Liu, Y.; Ulomsky, E.N.; et al. Abnormal Push-Pull Benzo[4,5]Imidazo[1,2-a][1,2,3]Triazolo[4,5-e]Pyrimidine Fluorophores in Planarized Intramolecular Charge Transfer (PLICT) State: Synthesis, Photophysical Studies and Theoretical Calculations. Dye. Pigment. 2022, 204, 110405. [Google Scholar] [CrossRef]
- Santos, M.N.B. External Heavy-Atom Effect on Fluorescence Kinetics. PhysChemComm 2000, 3, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Klemens, T.; Świtlicka, A.; Szlapa-Kula, A.; Krompiec, S.; Lodowski, P.; Chrobok, A.; Godlewska, M.; Kotowicz, S.; Siwy, M.; Bednarczyk, K.; et al. Experimental and Computational Exploration of Photophysical and Electroluminescent Properties of Modified 2,2′:6′,2″-Terpyridine, 2,6-Di(Thiazol-2-Yl)Pyridine and 2,6-Di(Pyrazin-2-Yl)Pyridine Ligands and Their Re(I) Complexes. Appl. Organomet. Chem. 2018, 32, e4611. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Room-Temperature | Low-Temperature (77 K) | ||||
|---|---|---|---|---|---|---|
| λabs, nm (ε, 103·M−1cm−1) | λPL, nm | ΦPL | τ, ns | λPL, nm | τ, ns | |
| n-Hexane | 364 (87.4), 291 (107.4) | 407 | 0.48 | 1.53 ± 0.01 | 438 | 1.48 ± 0.01 |
| Cyclohexane | 366 (57.0), 293 (68.8) | 408 | 0.54 | 1.55 ± 0.01 | 418 | 0.99 ± 0.06 (24.74%), 2.29 ± 0.02 (74.26%) |
| Toluene | 369 (74.6), 293 (96.7) | 435 | 0.64 | 2.55 ± 0.01 | 422 | 2.00 ± 0.01 |
| Chloroform [63] | 371 (74.6), 294 (100.2) | 487 | 0.84 | 4.30 ± 0.02 | 442 | 3.05 ± 0.01 |
| Ethyl acetate | 363 (74.1), 291 (92.2) | 468 | 0.70 | 3.57 ± 0.01 | 424 | 2.44 ± 0.01 |
| Tetrahydrofuran | 364 (75.2), 291 (101.8) | 467 | 0.75 | 3.81 ± 0.01 | 428 | 2.12 ± 0.01 |
| Dichloromethane | 369 (42.8), 292 (59.1) | 497 | 0.79 | 4.83 ± 0.01 | 439 | 1.78 ± 0.06 (41.12%), 3.43 ± 0.05 (58.88%) |
| Dimethylformamide | 367 (33.3), 291 (46.0) | 518 | 0.77 | 5.87± 0.02 | 439 | 1.61 ± 0.06 (30%), 3.46 ± 0.03 (70%) |
| Dimethylsulfoxide | 372 (46.8), 292 (67.4) | 528 | 0.83 | 6.56± 0.02 | 441, 516sh | 2.52 ± 0.01 |
| Acetonitrile [63] | 364 (31.6), 289 (41.5) | 527 | 0.63 | 5.53± 0.03 | 463 | 2.05 ± 0.20 (27.77%), 4.07 ± 0.12 (72.23%) |
| Methanol | 369 (47.3), 290 (66.6) | 557 | 0.02 | n.d. | 438 * | 2.95 ± 0.02, 0.75 ± 0.23 * |
| Butyronitrile | n.d. | 508 | n.d. | 4.89 ± 0.01 | 429 | 2.33 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maroń, A.M.; Cannelli, O.; Socie, E.C.; Lodowski, P.; Machura, B. Push-Pull Effect of Terpyridine Substituted by Triphenylamine Motive—Impact of Viscosity, Polarity and Protonation on Molecular Optical Properties. Molecules 2022, 27, 7071. https://doi.org/10.3390/molecules27207071
Maroń AM, Cannelli O, Socie EC, Lodowski P, Machura B. Push-Pull Effect of Terpyridine Substituted by Triphenylamine Motive—Impact of Viscosity, Polarity and Protonation on Molecular Optical Properties. Molecules. 2022; 27(20):7071. https://doi.org/10.3390/molecules27207071
Chicago/Turabian StyleMaroń, Anna Maria, Oliviero Cannelli, Etienne Christophe Socie, Piotr Lodowski, and Barbara Machura. 2022. "Push-Pull Effect of Terpyridine Substituted by Triphenylamine Motive—Impact of Viscosity, Polarity and Protonation on Molecular Optical Properties" Molecules 27, no. 20: 7071. https://doi.org/10.3390/molecules27207071